发布时间:2025-09-02 浏览次数:2664
近年来,sp3杂化的手性四配位硼化合物因其独特的结构和电子性质而受到广泛关注。由于硼原子半径小、缺电子的特性,构建稳定的硼立体中心手性分子存在较大挑战,通常需要借助路易斯碱或刚性骨架来稳定其四面体构型。目前,该类化合物的合成主要依赖于外消旋拆分或手性底物诱导的不对称合成策略,而催化合成硼中心手性化合物的方法直到最近几年才取得突破。然而,现有方法大多仍局限于具有环状螯合结构的硼络合物底物,以确保四配位硼产物在立体构型上的稳定性。因此,设计并合成结构稳定的前手性硼烷底物,已成为构建硼手性中心所面临的关键挑战。
深圳格拉布斯研究院双聘教授徐明华教授团队长期致力于铑(I)/手性烯烃配体及其参与的不对称催化反应研究,在铑(I)催化芳基卡宾、二芳基卡宾、烷基卡宾不对称B–H键插入反应研究中取得了一系列成果(J. Am. Chem. Soc. 2015, 137, 5268; Angew. Chem. Int. Ed., 2024, 63, e202412193; CCS Chem. 2025, 7, 2173–2184; Chem. Soc. Rev. 2025, 54, 6505–6524)。近日,徐明华教授团队报道了一项最新进展:以铑(I)/手性双烯配体和铜(I)/双噁唑啉为催化剂,N-杂环卡宾(NHC)取代的前手性二氢硼烷为底物,通过对映选择性去对称化B–H键插入反应,成功构建了含有碳/硼两个相邻手性中心的有机硼烷化合物,取得最高99%收率、99% ee以及93:7 dr。研究发现,铑催化剂与铜催化剂在二氢硼烷的B–H键插入反应中表现出相反的非对映选择性,通过金属与配体构型的合理组合,可以精准构筑碳、硼两个相邻立体中心,实现反应的立体发散性合成,得到所有四种立体异构体(图1)。
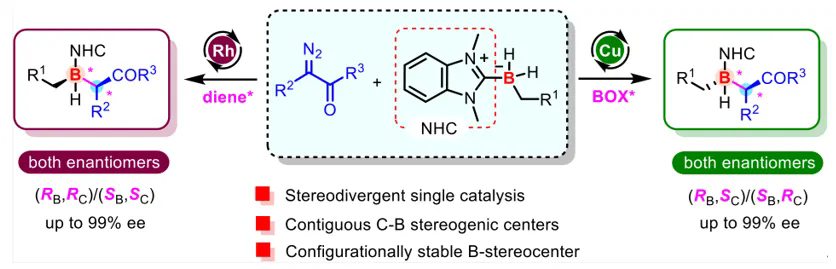
图1. Rh(I)/Cu(I)催化的卡宾B–H键插入反应
作者首先尝试了甲基吡咯及DMAP络合的取代硼烷,发现它们在铑/双烯催化体系下与芳基重氮酯不发生反应。随后,作者系统考察了一系列NHC络合的取代硼烷,发现该类硼烷表现出良好的反应活性与立体选择性。其中,以苯并咪唑衍生的卡宾硼烷络合物为底物,采用新型大位阻酰胺取代的双烯配体铑络合物([Rh(L5)Cl]2)作为催化剂时,反应取得了最佳结果(99% 收率,99% ee,93:7 dr)。值得注意的是,在铜/手性双噁唑啉催化体系中,产物的非对映选择性发生反转,表明该反应的立体选择性具有催化剂依赖性。经过对多种双噁唑啉配体的筛选,作者发现含有手性茚结构的配体L11能够以优异的对映选择性和非对映选择性实现目标对映异构体的合成。进一步的底物适用性研究表明,在Rh(I)和Cu(I)催化体系下,芳环上带有不同取代基的重氮酯和重氮酮类底物均可获得优异的结果(最高达97:3 dr和99% ee,部分例子如图2)。此外,该反应对多种取代的烷基NHC硼烷化合物也表现出良好的兼容性,大部分底物均可实现99% ee的对映选择性。
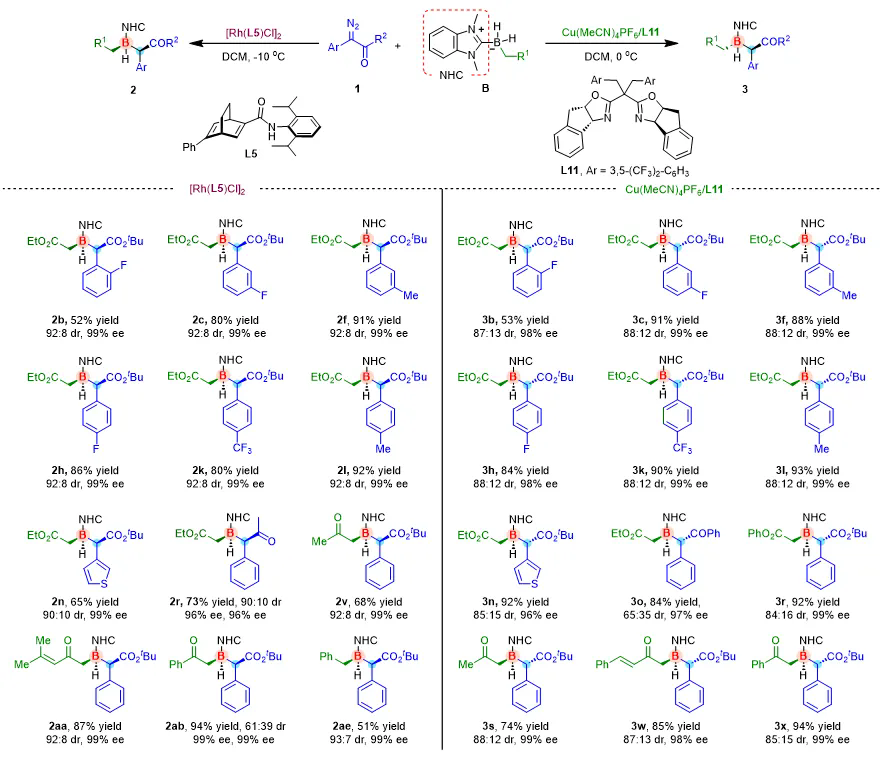
图2. Rh(I)/Cu(I)催化的卡宾B–H键插入反应底物普适性例子
作者利用不同金属催化体系可实现相反非对映选择性的特点,分别采用Rh(I)/(R,R)-L5、Rh(I)/(S,S)-L5与Cu(I)/(R,S)-L11、Cu(I)/(S,R)-L11催化二氢硼烷B5与苯基重氮叔丁酯1a的去对称化B–H键插入反应,以高收率和高立体选择性成功合成了四个立体异构体,实现了含碳/硼双手性中心有机硼化合物的立体发散性合成。作者进一步培养了这四个异构体的单晶,并通过X射线单晶衍射分析验证了它们的绝对构型(图3)。
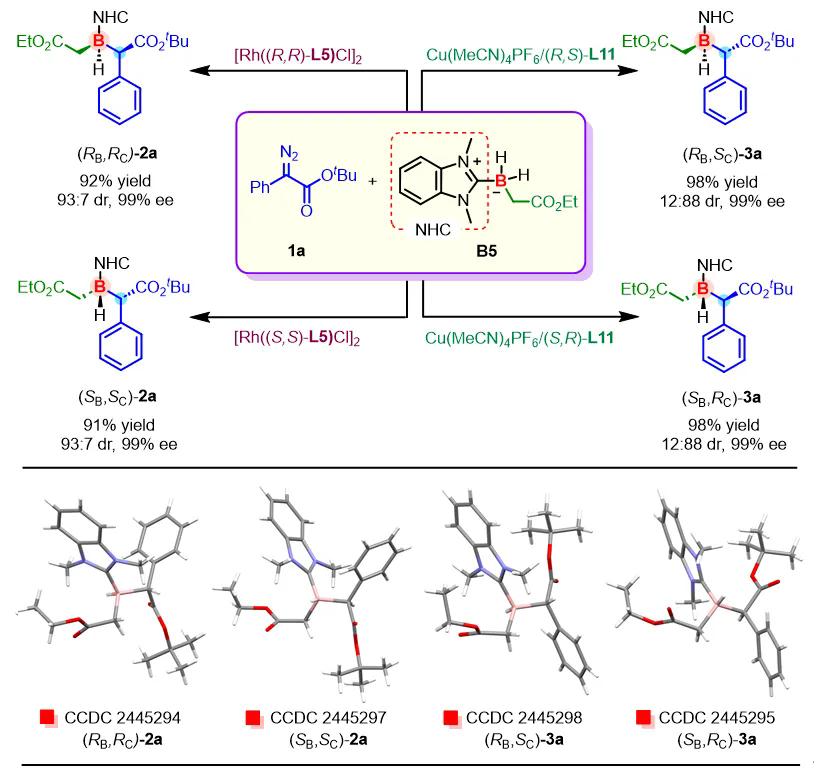
图3. 立体发散性合成
为评估所合成的手性有机硼烷的合成应用,作者对其进行了多种官能团转化实验。结果表明,该类手性NHC-硼烷化合物可在温和条件下实现多样转化:既可被还原为相应的二醇,也可在室温下发生氟代反应,生成四取代有机硼化合物。此外,通过苯酯的胺解反应及酯交换反应,可分别得到相应酰胺和香叶醇酯类衍生物。值得注意的是,在上述所有转化过程中,产物对映选择性均得以良好保持,表明该类手性硼中心在官能团化过程中具有优异的立体稳定性(图4)。
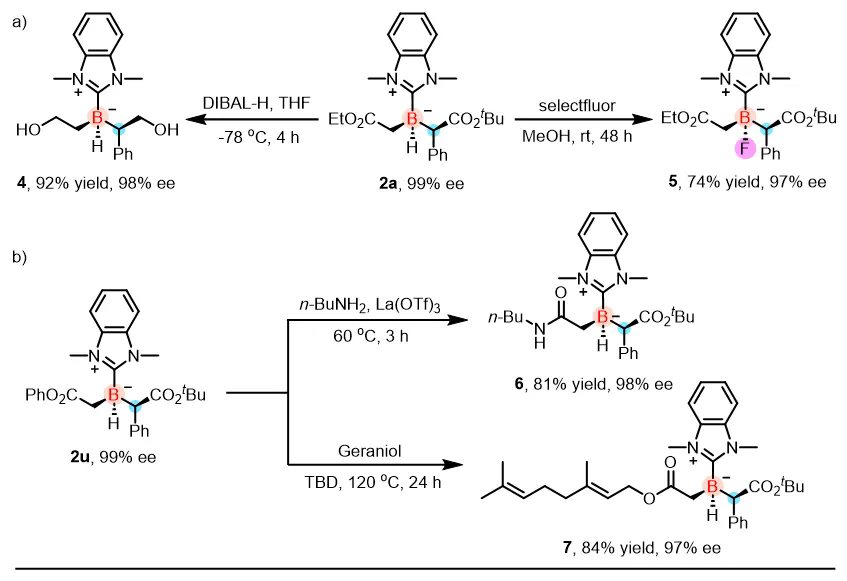
图4. 手性烷基硼烷的衍生化研究
为深入探究反应机理,作者开展了竞争性动力学同位素效应实验。通过研究重氮化合物1a与NHC-硼烷B5及其氘代类似物B5-d2在不同催化体系下的反应,测得在铑催化条件下的kH/kD值为2.03,而在铜催化下为2.23。这些结果表明,B–H键插入过程表现出明显的动力学同位素效应,进一步支持该反应通过协同反应机制进行。此外,相近的KIE值也说明反应机理在不同金属催化剂下保持一致,与金属种类无关(图5)。
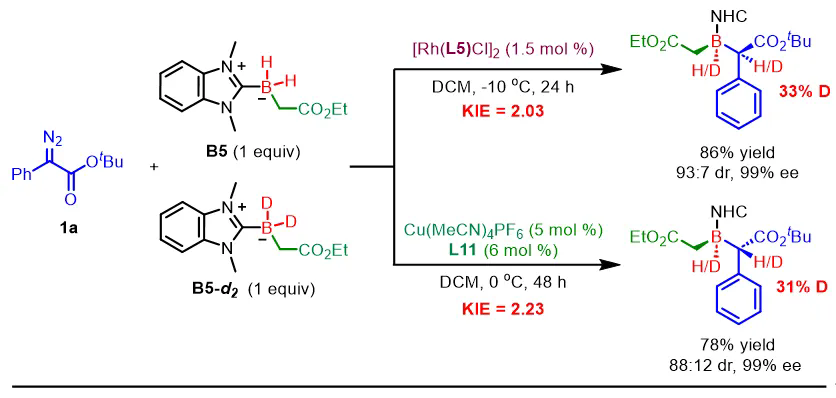
图5. 竞争性KIE实验
为解释非对映选择性差异的来源,作者采用DFT计算方法分别对铑(I)/手性双烯和铜/手性双噁唑啉催化的B–H插入反应过渡态进行了研究(过渡态结构见图6)。计算结果表明,在这两种催化体系中,卡宾中间体均通过手性配体所提供的立体环境识别前手性硼烷的空间构象,并以协同过渡态的方式选择性地插入至NHC-硼烷的某一特定B–H键中。在铑/手性双烯催化体系中,Rh-TS-1过渡态能量较低,其优势构象中硼烷的烷基取代基朝向远离卡宾的一侧,有利于B–H键通过三元环过渡态参与反应,最终生成(RB,RC)构型产物。而在铜/手性双噁唑啉催化体系中,Cu-TS-1过渡态更为稳定,其中硼烷倾向于从双噁唑啉配体空间位阻较小的一侧接近,同时其烷基取代基更易远离配体上边臂基团,从而促进(RB,SC)产物的形成。
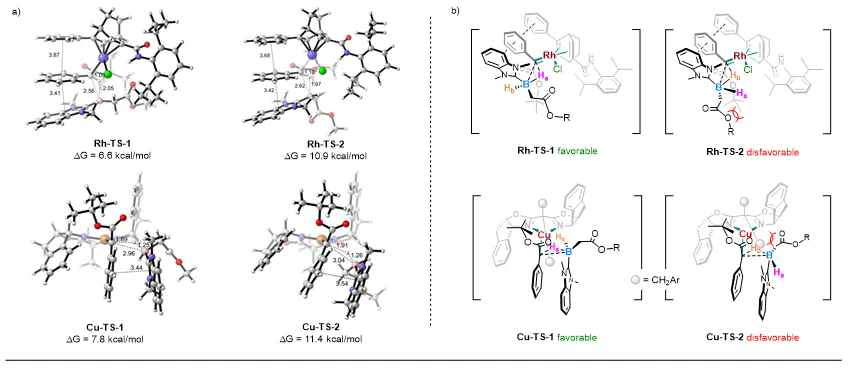
图6. DFT计算研究
四面体硼构型的稳定性不仅受配体与硼原子之间键合强度的影响,还与取代基的电负性和空间效应等多种因素密切相关,这些因素共同调控硼化合物的几何构型。为量化硼原子上取代基的空间排布与理想四面体构型之间的偏离程度,研究引入了基于键角计算的四面体特征参数(tetrahedral character, THC)。THC值越高,表明取代基的空间排布越接近理想四面体,相应的有机硼化合物也越稳定。基于单晶衍射数据,作者计算出手性硼化合物 (RB,RC)-2a的THC值高达91.1%,说明其结构已经非常接近正四面体杂化构型。这也是目前文献报道的THC值最高的手性硼化合物(图7)。值得强调的是,该化合物表现出极佳的稳定性:在常温下储存三年,或在100°C的甲苯中加热10小时后,均未发生分解或消旋现象。
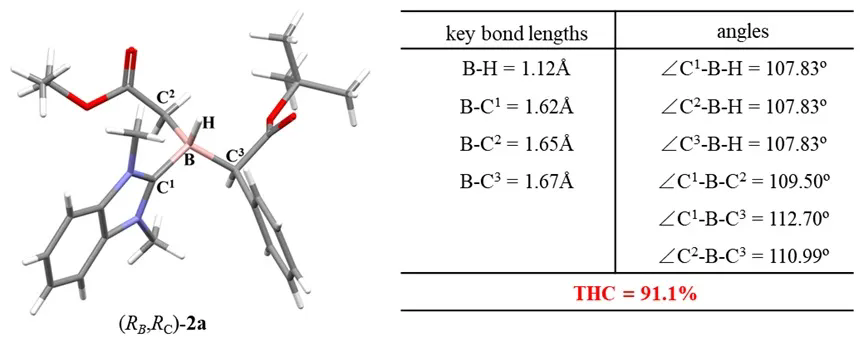
图7. (RB,RC)-2a的THC值
综上所述,徐明华教授团队以烷基取代的前手性NHC-硼烷和芳基重氮化合物为底物,分别在铑(I)/手性双烯配体和铜(I)/双噁唑啉两类催化体系下,通过高对映选择性的去对称化B–H键插入反应,成功实现了含连续碳/硼手性中心的有机硼烷化合物的立体发散性合成。通过合理选择两种手性催化体系,可系统性地获得目标产物的所有立体异构体。该反应对多种电性取代的芳基重氮底物以及不同烷基取代的氮杂卡宾硼烷均表现出良好的适用性,并以优异的反应效果(最高达99%收率、99% ee和93:7 dr)获得相应产物。所得手性有机硼烷化合物结构稳定,为后续衍生化研究提供了良好基础。此外,该研究为构建结构稳定的非环状NHC取代的硼中心手性分子提供了一条简洁高效的新路径,也为通过单一催化剂体系同时调控碳/硼两个手性中心的立体发散合成提供了重要案例。