发布时间:2024-07-14 浏览次数:8253
电化学作为一种绿色经济的电子转移的手段,通过电能输入,可以实现常规热化学无法实现的逆热力学反应。在这些逆热力学反应中,去消旋化是一种颇具潜力的构建手性中心的新颖手段,它能够在不改变化学结构的情况下,将一对外消旋体选择性地转化为单一对映体。尽管最近光催化去消旋化有诸多进展,实现电催化的去消旋化反应仍然是一项艰巨的挑战。2023年,清华大学罗三中教授在综述中展望到:“电催化及磁场催化,有潜力拓展去消旋化的边界。”(J. Am. Chem. Soc. 2023, 145, 10917)尽管备受期待,但电极反应不经过激发态化学,难以有效地正交进行正向反应与逆向反应,目前尚未有重大突破。最近,深圳格拉布斯研究院王健纯副教授团队成功利用了新颖的化学修饰电极策略,克服了不同催化剂氧化还原电势兼容性问题,实现了仲醇的电化学去消旋化反应,相关成果发表于Nature Catalysis上。
去消旋化反应实现的最大难点在于需要打破动力学上的微观可逆性。传统上,利用化学催化的方式,可以通过牺牲化学计量的氧化还原试剂将反应驱离化学平衡位置,但氧化剂和还原剂的兼容问题构成了巨大的挑战。尽管化学家们采用了多种时空分离策略来应对氧化还原兼容性这一挑战,但普适性有限。最近,研究者利用光催化的激发态与基态势能面分离的特性打破微观可逆性,通过能量转移或电子转移等新策略,可以实现去消旋化。激发态策略似乎是目前可以打破微观可逆性原理的独特例外,那么,如果不利用激发态势能面,是否还能打破微观可逆性呢?
作者认为:可以利用金属氢催化,并结合阳极氧化动力学拆分和阴极还原的模式实现所谓的循环去消旋化。为了打破微观可逆性,氧化和还原必须使用不同的催化剂。具体的设计思路是:在阳极通过手性催化剂M1+选择性将其中一种构型的底物氧化成酮,而M1-H则被重新氧化成M1+。(图1)而在阴极又将酮重新还原为外消旋的一对醇。这样即使拆分的选择性不高,也可以通过多次循环,逐渐提高e.e. 值。作者将此命名为电化学循环去消旋化反应(ECD)。
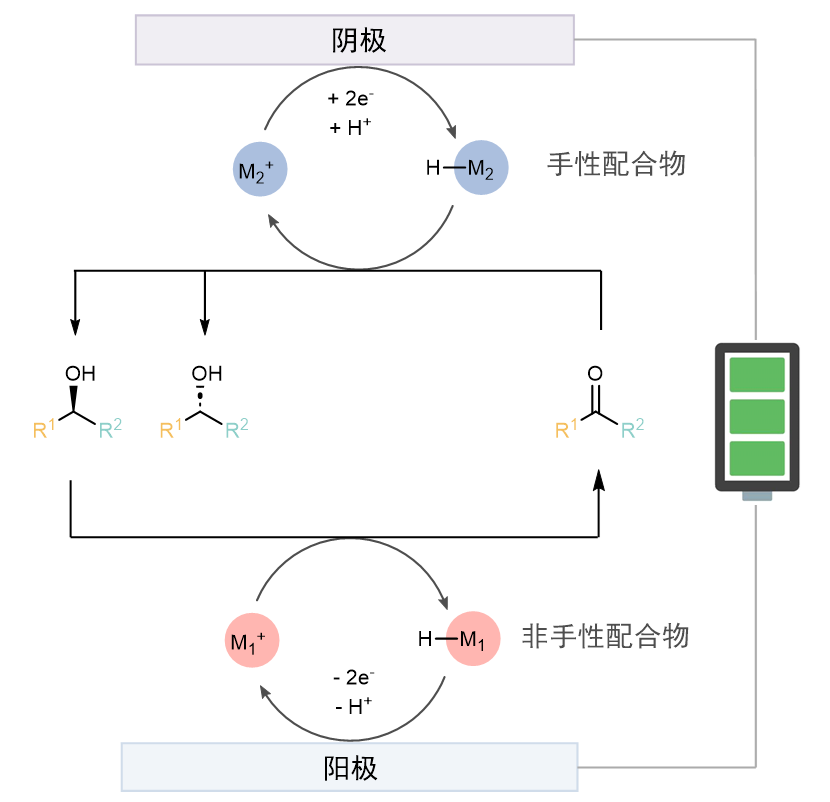
图1. 电化学循环去消旋化的初步设计
那么,该如何选择这一对关键的氧化还原催化剂呢?作者认为可以借助文献已知的物理化学参数来设计反应。首先,借氢反应的驱动力来源于酮和亚胺接受负氢(hydride)的能力不同。(图2)而电化学去消旋化的驱动力来源于两个金属氢催化剂的提供负氢的能力不同,而这一能力可以用负氢性(hydricity,∆GH-)来定量描述。因为负氢从醇的底物转移至负责氧化的催化剂M1,而负责还原的催化剂M2又可以将负氢转移到酮上重新生成醇,因此提供负氢的能力排序如下:M2−H > 醇 > M1−H,即负氢性∆GH-(M2−H) < ∆GH-(M1−H)。
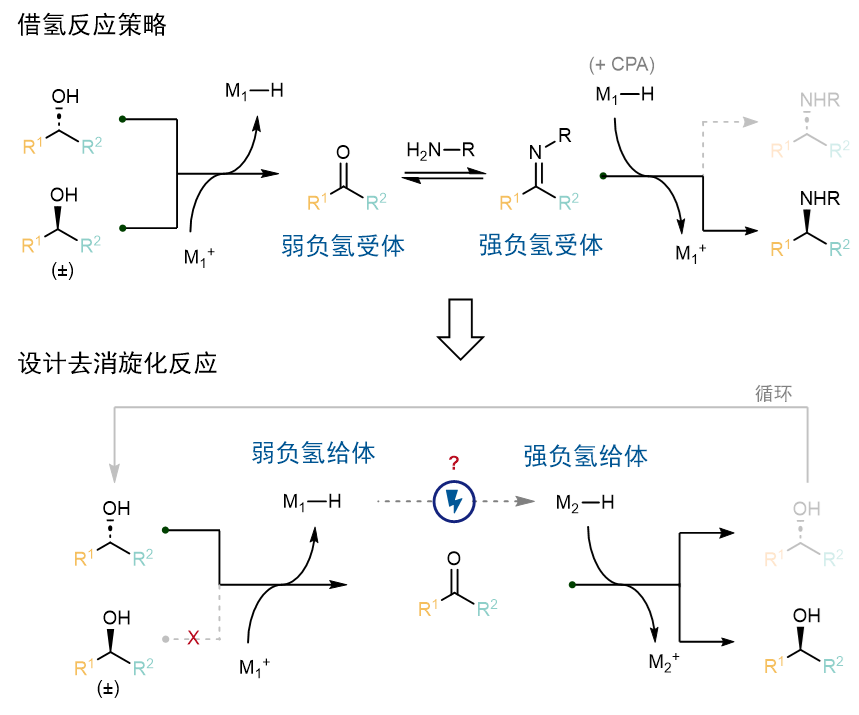
图2.电化学循环去消旋化与借氢反应的比较
其次,当电催化存在多个催化物种时,设计反应时还需要考虑不同物种氧化还原电势的兼容性。因为在阴极需要将电子优先转移给负责还原的催化剂M2+而不是负责氧化的催化剂M1+,即M2+需要优先被还原,E1/2(M1+/M1) > E1/2(M2+/M2)。因此,实现该反应的关键在于寻找到同时满足M2+更易被还原且M2−H提供负氢能力更强的一对催化剂!
可惜的是,这两个限制性要求无法同时被满足。事实上,在前人的研究中早已证明,无论金属类型或配体结构如何,金属氢物种的负氢性与其母配合物的还原电势之间存在很强的相关性。(ACS Catal. 2018, 8, 1313)这样的关联也不难理解:负氢等效于一个质子与两个电子,因此,M2−H作为一个强负氢给体,意味着生成负氢所需要的电子的能量更高,也就是说母配合物M2更难被还原。(图3)因此,克服氧化还原兼容性,也就是选择性地优先还原一个更难被还原的配合物,似乎是打破微观可逆性的关键。
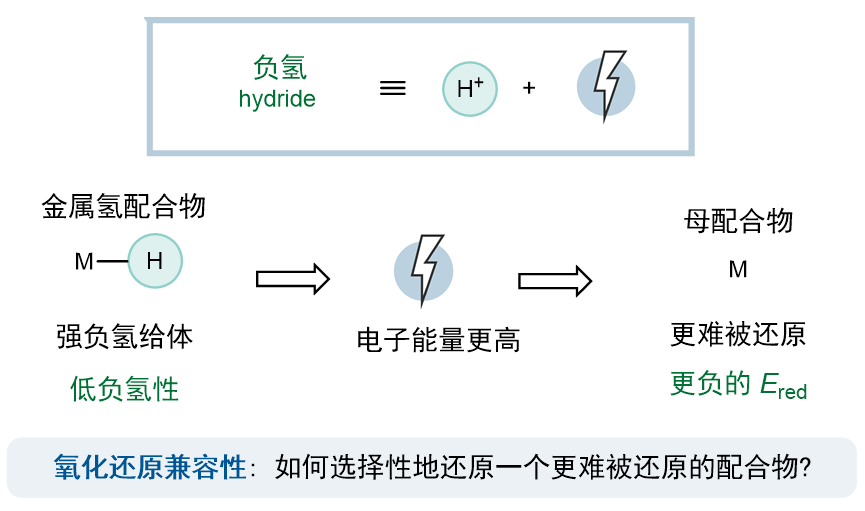
图3. 金属氢物种的负氢性与其母配合物的还原电势的关联性
接下来问题是,如何寻找一个普适的克服氧化还原兼容性的策略呢?与光化学虽然同为电子转移的手段,电化学的最大区别在于电子转移发生在电极表面,本质上是一个异相过程。能否充分利用电化学异相反应的特点呢?作者将目光投向化学修饰电极(CME)上。化学修饰电极通过共价或非共价作用将催化剂负载在电极表面,拥有降低过电势、降低载量、电极重复利用等优势。化学修饰电极在能源转化领域的应用较常见,但在有机合成领域却鲜有涉及,这或许是因为先前的应用并未带来新的反应性。作者设想,由于化学修饰电极表面催化剂浓度较高,即使该催化剂更难还原,它或许依然会被优先还原。于是,作者利用成熟的吡咯电聚合的方式将双联吡啶铑催化剂负载在碳毡电极表面,制成化学修饰电极Rh-CME。(图4)
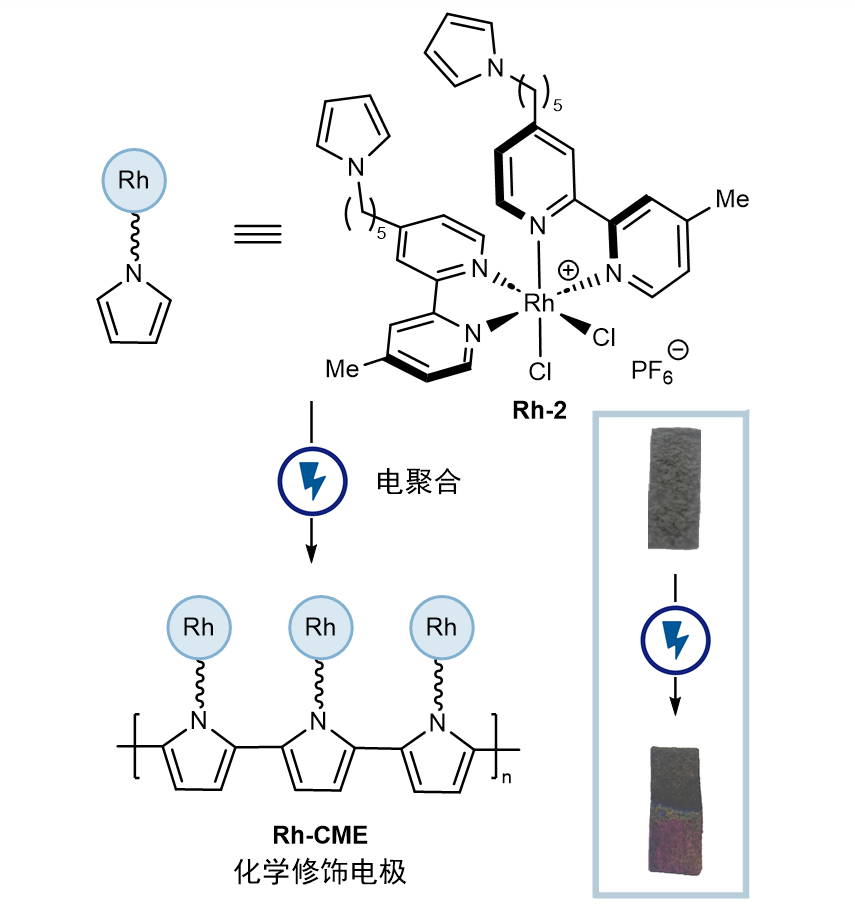
图4. 化学修饰电极的制备方法
先前,Noyori型催化剂被报道可以使用空气作为氧化剂实现氧化动力学拆分。作者发现在碱的作用下,通过电子转移-质子转移机理,可以成功利用电化学氧化实现仲醇的氧化动力学拆分。于是结合这些实验,作者利用铑配合物修饰的电极作为阴极,同时采用Noyori型催化剂作为阳极氧化的催化剂,成功实现了仲醇的电化学去消旋化。(图5)值得注意的是,该反应只能利用化学修饰电极Rh-CME实现,如果使用均相的铑配合物Rh-3则无法实现这一过程。
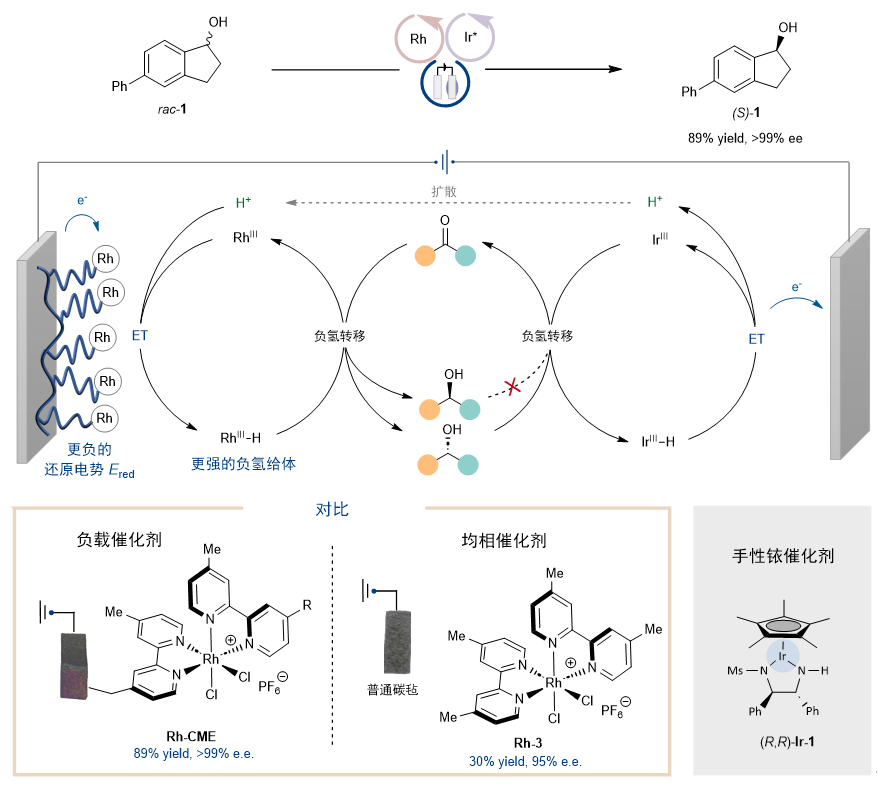
图5. 电化学循环去消旋化的实现,化学修饰电极与均相催化剂的比较
接下来,作者考察了电催化循环去消旋化反应的底物范围。(图6)该反应体现出宽泛的官能团兼容性,包括环氧、一级醇、硫醚、杂环等官能团均可兼容。各类底物,如环状的、非环状的仲醇均可适用,同时环状的芳基-芳基仲醇甚至环状的烷基-烷基仲醇也可以兼容。该方法也可以应用于一种强效酸性鞘磷脂酶抑制剂的中间体的合成。同时,氯雷他定、薄荷醇、核糖呋喃苷、香芹醇、香叶醇和奥司美芬衍生的仲醇也可以适用。
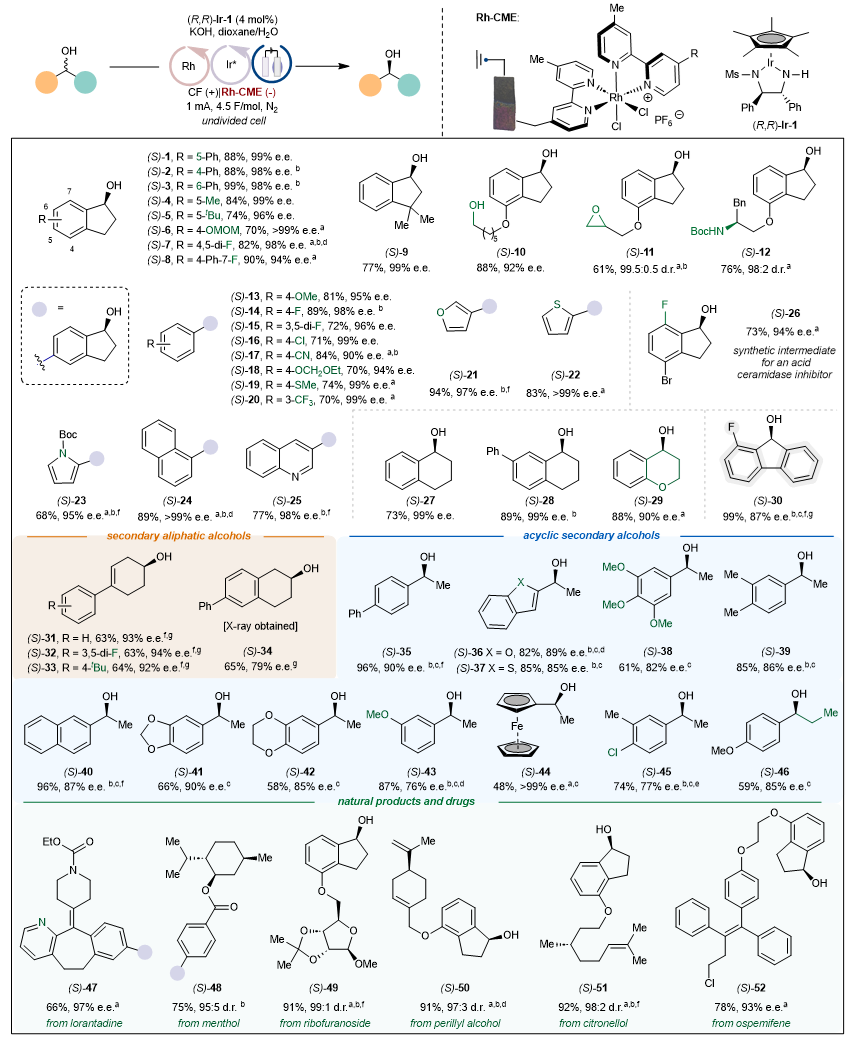
图6. 底物适用范围
作为该反应的应用,取代的 3-环己烯醇结构是各种生物活性化合物中的常见结构,然而,实现其对映选择性合成一直是个挑战,这可能是因为两边位阻差异较小。(图7)最近Brown课题组通过一个巧妙的立体选择性 [4+2]-环加成过程历经六步反应完成合成(R)-31,但需使用当量的手性烯基硼烷54。(Angew. Chem. Int. Ed. 2020, 59, 11432)对比而言,新方法提供了一种从易得的消旋体一步合成手性的3-环己烯醇的直接合成途径。除了步骤经济性和衍生化的便利性之外,新方法还可以方便合成两种对映体,而手性烯基硼烷试剂仅一种对映异构体相对易得。
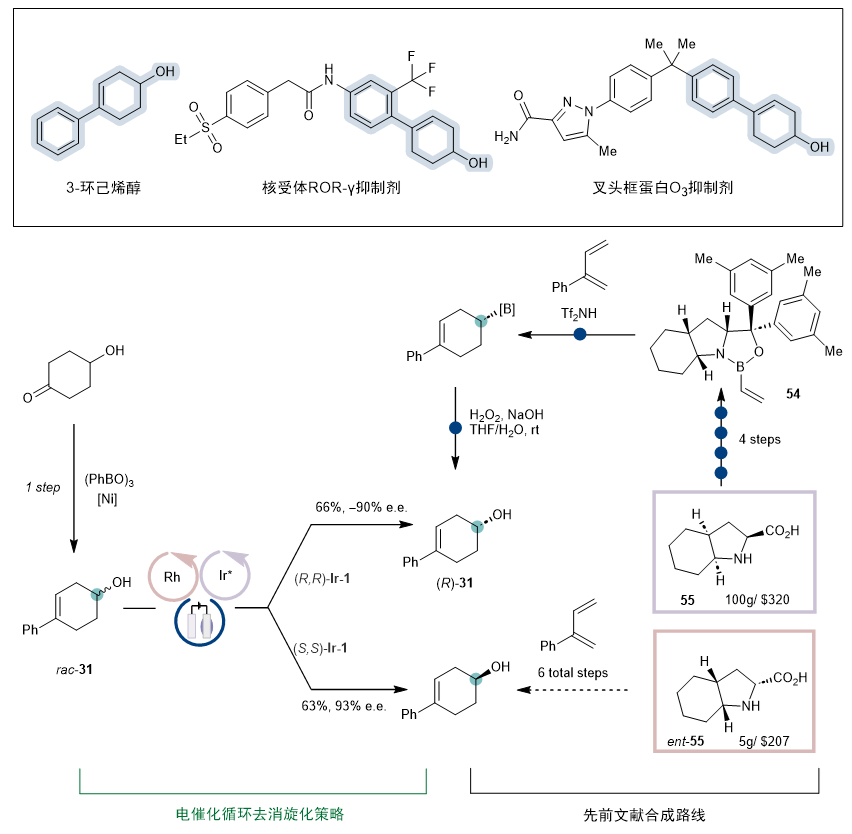
图7. 反应应用于手性3-环己烯醇的合成
值得一提的是,虽然是相同的手性铱催化剂,利用原文献(Angew. Chem. Int. Ed. 2008, 47, 2447)中空气氧化动力学拆分策略的底物适用性却相对窄许多。(图8)作者分析认为,即使一次循环拆分效果不佳(例如对于底物31,krel仅为4),通过多次循环拆分可以显著增加e.e.。同时,氧气作为氧化剂时会发生更多的副反应,而电氧化反应更具选择性。因此,新策略有望在不改变催化剂的前提下,将不成功的动力学拆分反应转变为成功的去消旋化反应。
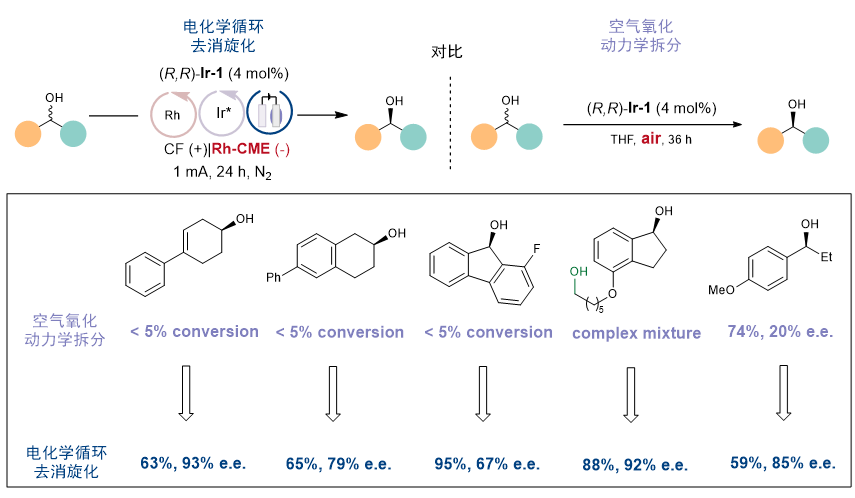
图8. 电化学循环去消旋化与文献中空气氧化动力学拆分的对比
有趣的是,该电催化去消旋化还表现出优异的化学选择性,可以选择性地对环状仲醇进行去消旋化,而不影响链状仲醇。因此,该方法提供了一种独特的途径来获取具有两个不同立体构型的二醇,而这种化合物用不对称氢化合成起来颇具挑战。(图9)举例来说,尽管经典的 CBS 还原可以方便地直接从二酮58合成(R,R)-和 (S,S)-构型的二醇57,但从二酮58合成(R,S)-和 (S,S)-构型的二醇57则难以实现。然而,通过该方法对环状仲醇进行差向异构化,可以成功获得(R,S)-和 (S,S)-构型的二醇57。
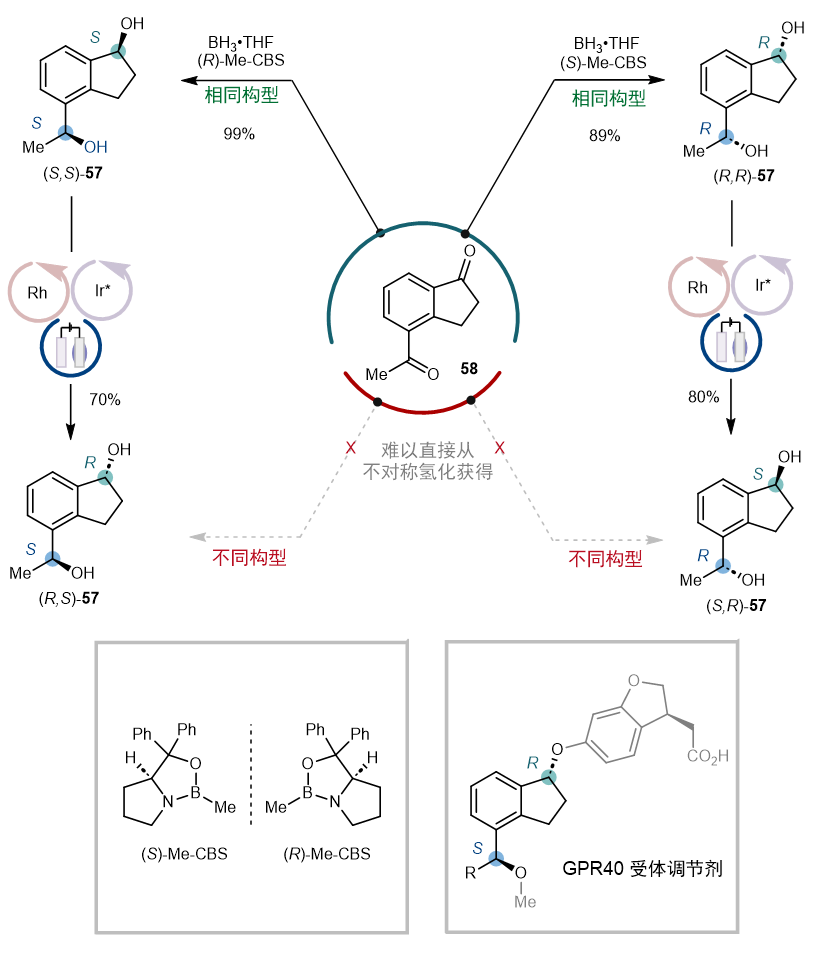
图9. 化学选择性带来的选择性立体中心翻转
机理实验方面,首先,作者通过循环伏安实验证明了氧化还原不兼容:和先前研究一致,RhIII比IrIII更难被还原,且Ir−H比还原态的Rh更难被氧化。(图10)这也证明通过化学修饰电极,可以使得难以被还原的RhIII优先被还原,同时,也可以防止还原态的Rh在阳极被氧化。其次,通过氘代实验,作者证明了Rh−H的来源是质子的还原,而非从Ir−H的负氢转移。
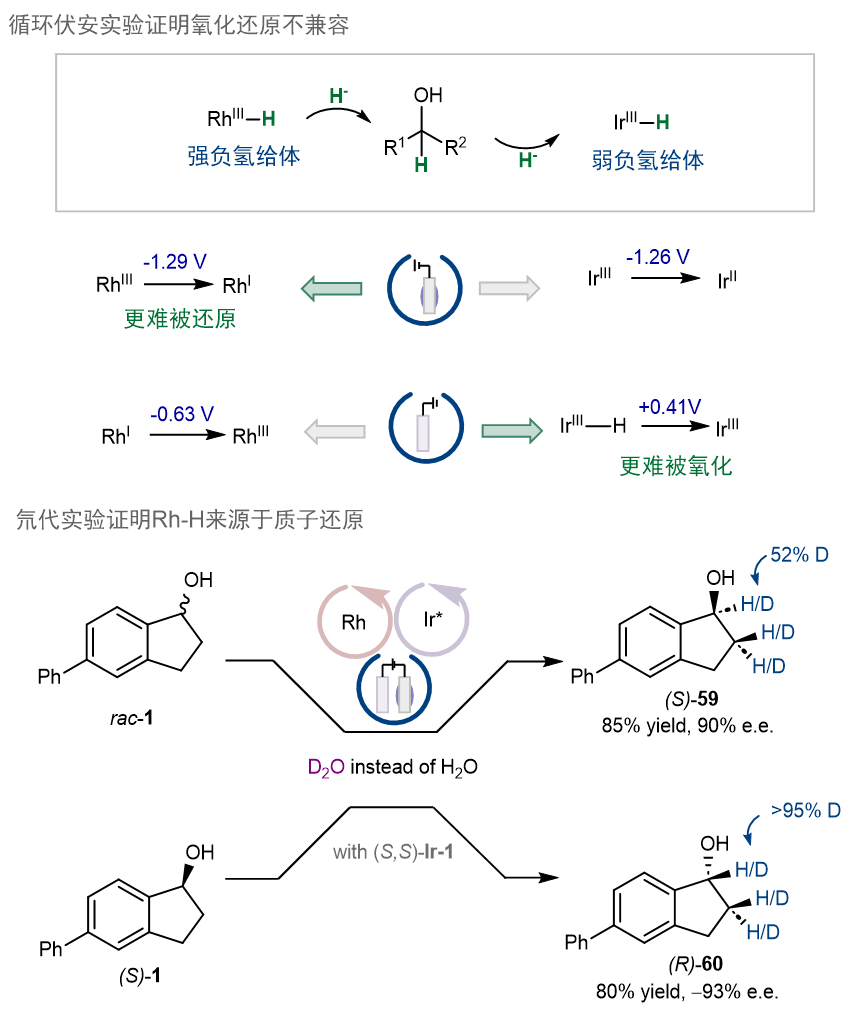
图10.循环伏安实验及氘代实验
该反应也具有化学修饰电极所具有的一般优势。第一,电极可以重复利用。可以简单溶剂冲洗即可下一次实验使用。(图11)这也与SEM实验结果符合,反应后Rh仍存在于电极表面,同时也说明了聚合物网络可以防止不稳定的低价铑失活。第二,催化剂载量低。通过电化学实验及ICP-MS实验可以测量出2.0 mmol反应的催化剂载量仅为0.05 mol%左右。可回收与低载量的特性也让化学修饰电极的使用增加了实用性。

图11. 电极的表征、重复利用实验及载量测量实验
总结
在本工作中,王健纯副教授团队成功实现了电化学循环去消旋化反应。通过鲜有涉足的化学修饰电极,巧妙克服了氧化还原兼容性问题,从而打破了微观可逆性原则。该方法有望为其他逆热力学电催化反应提供了一种新思路。该工作以“Electrocatalytic Cyclic Deracemization Enabled by a Chemically Modified Electrode”为题发表在Nature Catalysis上。此工作南方科技大学为唯一通讯单位,王健纯副教授为通讯作者, 22级博士研究生祝呈捷为第一作者,硕士研究生杨秀莹为第二作者。上述工作得到科技部国家重点研发计划、国家自然科学基金委员会、广东省催化化学重点实验室、深圳市科创委的经费支持。